
The Dovetail® Analysis Portal is available to all Dovetail Genomics customers and enables the streamlined analysis
of NGS FASTQ files generated from Dovetail® linked-read libraries. Supported workflows include
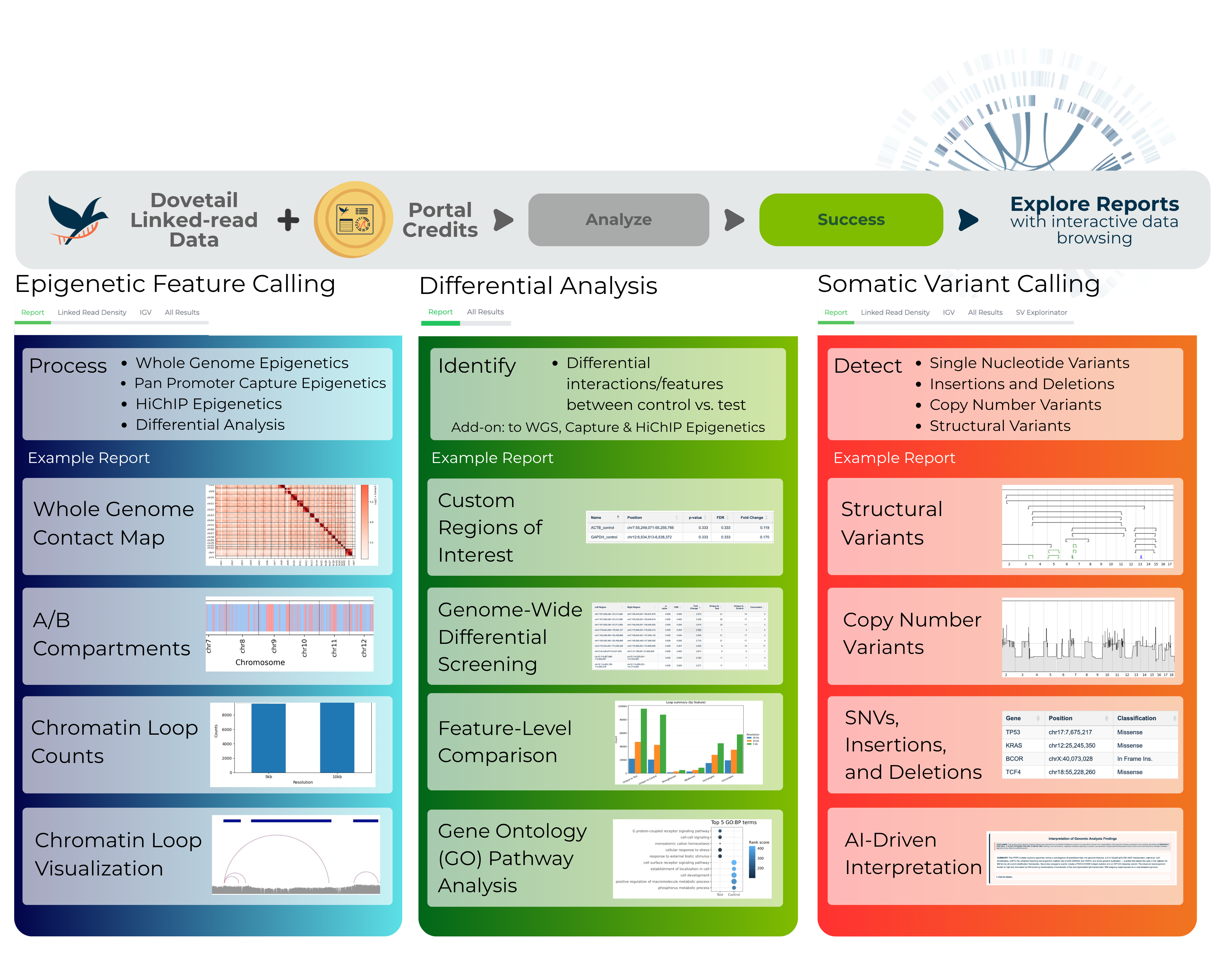
1. Somatic variant calling (human only)
2. Epigenetic feature calling (human and mouse only)
3. Differential epigenetic analysis (human and mouse only)
The portal delivers results through a summary report and a packaged,
easily downloadable archive. In addition, it offers interactive data browsing directly through the portal.
Each analysis requires a Dovetail® Analysis Portal credit. One credit for files up to 60GB in total and two credits for files 61GB-100GB in total. If your files are larger than 100GB please contact support@cantatabio.com.
Analysis pipelines are fully supported and maintained by Dovetail Genomics. Should you have any inquiries, please refer to our FAQ below or contact support@cantatabio.com.
NEW!!! We support inter-AWS account file transfers for data upload and results delivery. Data delivered to your personal AWS S3 are not subject to the 30-day storage time through our portal. Please see our General FAQ for more information.
Q: Is there a specific browser I should use for the Dovetail® Analysis Portal?
Yes, the Dovetail® Analysis Portal should be accessed using Chrome.
Q: Do I need bioinformatics training to use the portal?
No. The portal is designed to be user-friendly and does not require command-line knowledge. Analysis can be performed through a few simple button clicks.
Q: Can I provide metadata information?
No. The Dovetail® Analysis Portal is not HIPAA-compliant. Please make sure your file (including filename) contains no protected health information or personally identifiable information.
Q: Can I use the Dovetail® Analysis Portal for diagnosis?
No. The Dovetail® Analysis Portal is for Research Use Only and not for use in diagnostic procedures on patients.
Q: I set up my account, but I'm not able to upload data or submit an analysis. Why?
Credits are required to upload or analyze your own data. However, after signing up for an account, you can explore the portal and review publicly available example datasets and results.
Available demo data:
Q: How do I purchase portal credits?
Credits are available by contacting your sales representative or by purchasing directly from our webstore. Credits are added to your account once payment has been received. If you've already purchased credits but don't see them reflected in your account, reach out to support@cantatabio.com.
Q: How many credits are required for one analytical run on the Dovetail® Analysis Portal?
A single credit covers one analytical run of up to 60 GB (typically ~30X genomic coverage for gzipped FASTQ files) of total FASTQ data per sample (combined read 1 + read 2 files). Data sets between 60-100 GB (~80X genomic coverage) require two credits.
Q: When are credits deducted from my account?
Credits are deducted only upon the successful completion of an analysis run. If incorrect input files are selected and the run completes successfully, a credit will still be deducted. Please ensure that you are selecting the correct input files before submitting your analysis.
Q: What happens if my run fails? When is it appropriate to use the RETRY button?
If a run fails, you can retry it up to two times using the RETRY button. If the run is still unsuccessful after two attempts, please contact support@cantatabio.com for assistance. Retrying a failed run does not deduct additional credits from your account.
The RETRY button can be used if a run times-out. Analysis timeouts can happen during computationally heavy processes or data transfer post analysis. If your run fails to display results and complete, you can try to resume it through the RETRY button. The RETRY button does not allow you to reconfigure your run.
Q: Are credits refundable?
No, credits are non-refundable.
Q: How do I upload data to the Dovetail® Analysis Portal?
Data can be uploaded directly from your computer to the portal. For datasets larger than 60 GB, contact support@cantatabio.com to arrange an SFTP transfer, recommended for larger datasets. If you encounter upload issues, contact support@cantatabio.com.
If your FASTQ files are stored in your AWS S3 bucket, you don't need to upload the files to the portal. Instead, add the S3 path in the 'sync' field at the top of the 'Files' page to connect your bucket to the portal. Make sure your S3 bucket has the correct permissions, so the portal can access your files —detailed instructions are provided here.
Q. What input files are needed to run the analysis?
You will need the R1 and R2 FASTQ files generated from sequencing a Dovetail® LinkPrep™, Micro-C, HiChIP, or promoter capture library. The portal currently supports human samples only for Somatic variant calling and human and mouse samples only for Epigenetic feature calling.
Q: What FASTQ naming convention is acceptable?
Our portal will identify read 1 and read 2 using “R1” and “R2” regex. Be sure your FASTQ file names contains the character string R1.fastq.gz and R2.fastq.gz per paired file name.
For example, the following file name formats are acceptable:
DTG_microC_R1.fastq.gz and DTG_microC_R2.fastq.gz
DTG_microC_R1_001.fastq.gz and DTG_microC_R2_001.fastq.gz
Q. My sample has four R1 and R2 FASTQ files? How can I upload these to the portal?
If your 30X sequencing sample is split across multiple sequencing lanes or runs and results in multiple FASTQ files, there is no need to combine them before upload. You can upload the files separately—just be sure to select all relevant files when submitting the FASTQs for analysis.
Q: My FASTQ pairs are >60GB even though they are supposed to be ~ 30X coverage. What do I do?
Make sure your files are gzipped before upload to compress their file size. Gzipped FASTQ files typically have the following extension: fastq.gz. You can use the following command to gzip the file:
gzip file.R1.fastq
gzip file.R2.fastq
Q: How long is my data stored on the portal?
FASTQ files are stored for 30 days from the upload date. Analysis result files are available on the portal for 30 days after the run is completed, but may be downloaded for future analyses and viewing outside the portal. Data delivered to your personal AWS S3 are not subject to the 30-day storage time through our portal (see below for more information).
Q: How do I download my analysis result files once it’s completed?
Q: How do I use my own s3 bucket for storing result files?
{
"Version": "2012-10-17",
"Statement": [
{
"Effect": "Allow",
"Principal": {
"AWS": [
"arn:aws:iam::100977841808:root",
]
},
"Action": [
"s3:PutObject",
"s3:ListBucket",
"s3:GetObject"
],
"Resource": [
"arn:aws:s3:::BUCKET_NAME/*",
"arn:aws:s3:::BUCKET_NAME"
]
}
]
}
[
{
"AllowedHeaders": [
"Authorization",
"x-amz-date",
"x-amz-content-sha256",
"content-type"
],
"AllowedMethods": [
"GET"
],
"AllowedOrigins": [
"https://portal.cantatabio.com"
],
"ExposeHeaders": [
"ETag",
"Location"
],
"MaxAgeSeconds": 3000
}
]Q: How do I cite the analysis in my publication?
Please refer to the Appendix section at the bottom of the report that is included in the deliverables for method and tool citation.
Q. I purchased Dovetail Analysis Portal Credits for variant analysis. How do I submit a variant analysis run through the portal?
Submitting a variant analysis through the Dovetail Analysis Portal is quick and easy. Just follow these steps:
Q. My sequencing file is > 60GB, can I down-sample (sub-sample) it to run it through the 30X analysis workflow?
Yes, you can down-sample your FASTQ files before upload to perform 30X analysis. FASTQ pairs 60-100GB will be charged 2 credits. However, keep in mind certain samples (i.e. highly heterogenous or low purity) and/or low VAF variant detection will benefit from higher sequencing depth.
You can use the following approach to subsample the data:
First, install seqtk by following the instructions here: https://github.com/lh3/seqtk
After seqtk is installed, then run the following commands to subsample 400M read pairs from the full FASTQ files:
seqtk sample -s100 full.R1.fastq.gz 400000000 | gzip -c > subsample.R1.fastq.gz
seqtk sample -s100 full.R2.fastq.gz 400000000 | gzip -c > subsample.R2.fastq.gz
Q. I did not use a Dovetail® Kit to generate my libraries, and I'm interested in somatic variation detection. Can I still use the Dovetail® Portal for the analysis?
While the Dovetail® Analysis Portal can be used for somatic variation detection with data not generated using the LinkPrep™ or Dovetail-FFPE™ Kit, we cannot guarantee the success of the analysis run or provide support for results generated from non-Dovetail data and is therefore not recommended.
Q: What deliverables will I receive from a variant analysis run?
Deliverables from a Dovetail® Variant Analysis run include:
Q: What happens if my run fails?
If a run fails, you can retry it up to two times using the RETRY button. If the run is still unsuccessful after two attempts, please contact support@cantatabio.com for assistance. Retrying a failed run does not deduct additional credits from your account.
Q: How do I prioritize SV call quality?
High quality SV calls tend to: 1. Have high read support at high resolution. 2. Generate refined breakpoints 3. Contain features that can be confirmed by IGV or Linked Read Density Plots. Be suspicious of low-resolution calls (i.e. 1Mb, sometimes 100kb). These false positive (FP) calls tend to have lower scores and should be manually checked either in IGV or via Linked Read Density Plots.
Q: The purity solution output by the analysis portal does not match my pathology report. What should I do?
While modest differences are to be expected between any estimate of tumor purity, if the pathology-based purity estimate is substantially higher (e.g., more than 20%) than the bioinformatics-based estimate provided on the report, you can increase the minimum purity setting to align with the estimate from pathology. For instance, if the Dovetail analysis estimates a sample’s tumor purity to be 30%, but its estimate from pathology is 80%, an increased minimum purity threshold of 0.5 (50%) or even 0.7 (70%) may help guide the tool to identify an improved purity solution. Furthermore, if a sample is known a priori to have very high purity (e.g., cell line), then it is suggested that all analyses are run with a minimum purity threshold of at least 0.8 (80%).
Q: How do I cite the analysis in my publication?
Please refer to the Appendix section at the bottom of the report that is included in the deliverables for method and tool citation.
Q: What epigenetic feature analyses does the portal offer?
The portal offers the following epigenetic feature analyses:
Q. What Epigenetic Analyses does my data enable?
| Data Type | Genomic Coverage | # Read Pairs(2 x 150bp) | # of Libraries per sample | Epigenetic Analysis | Optional Add-on | Topological Feature | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Whole Genome data |
|
|
|
Whole Genome Epigenetics | Differential Analysis |
|
||||||||
| Pan Promoter Capture | N/A | 150M-300M | 1 | Pan Promoter Capture Epigenetics | Differential Analysis | Loop calls | ||||||||
| HiChIP Data | N/A | 300M | 2 | HiChIP Epigenetics | Differential Analysis | Loop calls |
Q: I purchased Dovetail Analysis Portal Credits for epigenetics feature analysis. How do I submit an epigenetic analysis run through the portal?
Submitting an epigenetic analysis through the Dovetail Analysis Portal is quick and easy. Just follow these steps:
Q: My sample/library was split across multiple sequencing lanes and resulted in multiple FASTQ files, how do I submit the FASTQ files for my sample?
If your sample was split across multiple sequencing lanes and resulted in multiple FASTQ files, there is no need to combine them before uploading. The portal allows you to select up to 4 sets of FASTQ files for a single sample - just be sure to select all the files corresponding to that sample/library before submitting the analysis.
Q: I prepared multiple Dovetail libraries to support 80X WGS sequencing for my sample. How do I upload my multiple libraries?
If you prepared multiple libraries to achieve 80X coverage for your sample for WGS epigenetics feature calling, the portal allows you to add up to 4 libraries per sample in a single run. Click “Add Library” to select the FASTQ files for each additional library. Once all libraries for that sample have been added, click “Finish” and proceed to submit the analysis.
Q: How do I view my analysis results?
Once the analysis is complete, you will receive an email notification. Log in to your account on the portal, then click on the “Analyze” tab in the top right corner of the page to access the results and explore the interactive features.
Q: What deliverables will I receive from a run?
Whole Genome Epigenetics
| File description | File Format(s) |
|---|---|
| Matrix files | .hic, .cool, .mcool |
| Alignment files | .bam, .bai |
| Valid pairs files | .gz, .px2 |
| Pairtools stats file | .csv |
| Fan-C AB compartment calls | .ab, .bed Indexed files for IGV: .bed.gz, .bed.gz.tbi, .bedgraph.gz, .bedgraph.gz.tbi |
| Arrowhead TAD calls (5, 10, 25kb) | .txt Indexed files for IGV: .bed.gz, .bed.gz.tbi |
| Mustache loop calls (5,10kb) | .tsv Indexed files for IGV: .bedpe.gz, .bedpe.gz.tbi |
| Hiccups loop calls (5, 10kb) | .bedpe |
| Whole genome epigenetics report | .html |
Pan Promoter Capture Epigenetics
| File description | File Format(s) |
|---|---|
| Matrix files | .hic, .cool, .mcool |
| Alignment files | .bam, .bai |
| Pairtools stats file | .csv |
| Valid pairs files | .gz, .px2 |
| Enrichment stats file | .txt |
| Chicago analysis files (10, 20kb) | .ibed, .txt, .png |
| Capture epigenetics report | .html |
HiChIP Epigenetics
| File description | File Format(s) |
|---|---|
| Matrix files | .hic, .cool, .mcool |
| Alignment files | .bam, .bai |
| Pairtools stats file | .csv |
| Valid pairs files | .gz, .px2 |
| FitHiChIP loop calls | .ibed, .txt, .png(packed in .tar.gz) |
| HiChIP epigenetics report | .html |
Q: What happens if my run fails?
If a run fails, you can retry it up to two times using the RETRY button. If the run is still unsuccessful after two attempts, please contact support@cantatabio.com for assistance. Retrying a failed run does not deduct additional credits from your account.
Q: The IGV is not displaying any data. What should I do?
Make sure to zoom into a specific chromosome for IGV to load and display the data.
Q: I purchased Dovetail Analysis Portal Credits for differential epigenetics analysis. How do I submit a differential epigenetic analysis run through the portal?
Submitting a differential epigenetic analysis through the Dovetail Analysis Portal is quick and easy. For detailed instructions, refer to the tutorial video above. The key steps are summarized below:
Once the analysis is complete, download the result files to your computer and explore the interactive features.
Q: What differential epigenetic analyses does the portal offer?
All differential epigenetic analyses are available as add-ons to the epigenetic feature analysis. This means you can only run a differential analysis AFTER completing the corresponding epigenetic feature analysis for your data type.
Differential analysis is performed at both a feature-level (i.e., for a given loop, is there a difference in call strength between test and control sample?) and an interaction signal-level (i.e., for a given bait, is there a difference in interactions between test and control sample?)
The portal offers the following differential analysis workflows:
Q: Can I compare between more than two experimental conditions?
No, the differential analysis is limited to two conditions/groups at a time.
Q: How many samples do I need for each group to perform differential analysis?
Q: Should my groups have the same number of samples?
Whole genome differential analysis supports only one sample in each group. For differential capture and HiChIP analyses, it is encouraged, but not required, to have an equal number of samples in each group.
Q: How does the order of the control or test groups affect my differential analysis output?
The direction of the fold change depends on this group assignment — reversing the group labels will invert the sign of the fold change and swap the associated values between the groups.
Q: I would like to run differential analysis pipeline, but I cannot find the FASTQ files of my samples in the selection drop-down menu. What should I do?
You can only run a differential analysis AFTER completing the corresponding epigenetic feature analysis for each sample in your differential analysis. Ensure that the epigenetic feature analysis for all samples in your differential analysis has successfully completed. When setting up the differential analysis, you will select the completed epigenetic feature run name as input for each sample (instead of a set of FASTQs).
Q: I have different sequencing depths between my conditions, can I still run differential analysis?
Yes. The differential pipelines perform normalization that accounts for library-specific biases, such as differences in coverage. Samples/conditions sequenced below Dovetail’s recommended depth may underperform.
Q: What happens if I mislabel samples?
You will likely need to re-run your analysis. Reach out to our support team (support@cantatabio.com) for assistance. Please note that credits used for the mislabeled run will not be refunded.
Q: What is a region of interest BED file and how is it used in the differential analysis?
A region of interest (ROI) BED file is an optional input that defines specific genomic regions you want to focus on during the analysis. It is a simple text file that lists chromosome coordinates (e.g., chromosome, start, end, regionID) for each region. When provided, the differential pipeline performs an additional calculation describing the features unique or shared between groups for each region. You will also receive example visualizations of the interaction matrix for each region. This is useful if you want to focus on particular loci—such as known genes, regulatory elements, or previously identified regions—without being overwhelmed by genome-wide results. This analysis is performed by taking the KR-normalized matrix files and extracting a fixed window around each region , then summarizing the interaction signal within that window by averaging log-transformed contact frequencies across a defined off-diagonal distance band. These per-sample scores are compared between groups to calculate a fold-change, statistical significance, and FDR, providing a quantitative measure of how chromatin structure differs at each region of interest. While your BED file can have as many entries as you like, the report captures top 100 differentially interacting regions ranked by fold change. You can find all the output for the regions of interest you submitted in the roi_annotated_with_features.tsv file included in the Deliverables.
Example BED file (Tab delimited):
chr8 127733433 127744951 MYC
chr11 67375961 67659024 special_region_1
Q: What deliverables will I receive once the run is completed?
For each differential analysis run, you will receive a summary HTML report file, as well as accompanying results files. We recommend that you begin by reviewing the report and then proceed to the other deliverables.
Differential Analysis
| File description | File Format(s) |
|---|---|
| Summary report | .html |
| Unique/Concordant feature list | .txt |
| Top differential region plots | .tar.gz file of .png images |
| Region of interest plots | .tar.gz file of .png images |
| Raw bin-level differential results | .txt |
| GO Ontology analysis results | .txt |
Q: What happens if my run fails?
If a run fails, you can retry it up to two times using the RETRY button. If the run is still unsuccessful after two attempts, please contact support@cantatabio.com for assistance. Retrying a failed run does not deduct additional credits from your account.
Q: Why are my results empty or nearly empty?
This can occur in certain scenarios and usually indicates poor library quality. Please review the QC metrics for each sample. Reach out to our support team (support@cantatabio.com) for assistance.
Q: How do I cite this analysis workflow and where can I get additional details on the analysis methods for the Dovetail differential workflows?
Refer to bottom of report and Differential readme.doc